Since studies related to COVID-19 are attracting great scientific interest, we recently focused our activities in recovery of viral sequences from swab samples by two methods: amplicon-Seq and RNA-Seq (using Trio RNA-Seq library preparation kit by Tecan Genomics as reported in our previous post).
Despite the amplicon-Seq limitations, the technique can offer, with adequate optimization, the most cost-effective way to access viral genome sequences on a massive scale to purse effective monitoring of the virus genetic diversity and spread patterns. We performed the analysis of swab samples on the Illumina sequencing platform by the direct amplification with tiled multiplexed primers, as suggested by the ARTIC protocol. By slightly decreasing the annealing temperature and modifying the sample amplification step, we were able to recover the major amplicon dropouts.
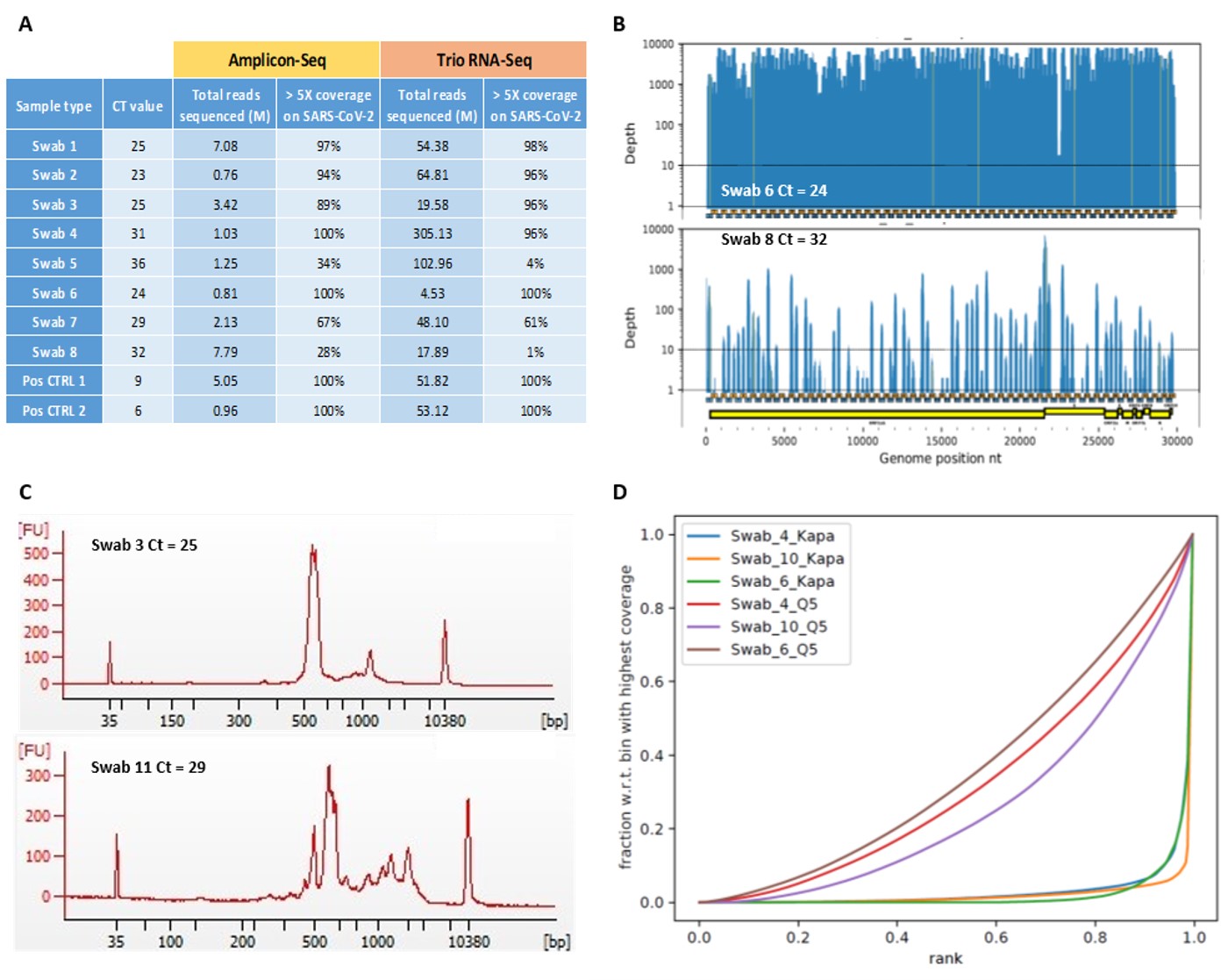
Since primer pools were missing Illumina tails, amplicons were converted to Illumina-compatible libraries by Celero™ DNA-Seq kit (Tecan Genomics) and sequenced on the MiSeq platform in 300bp PE mode. The sequencing depth and the percentage of SARS-CoV-2 genome with a minimum 5X coverage for samples processed with both amplicon-Seq and RNA-Seq approach is shown in the table below (Figure A). The results were correlated across the two methods and, for both procedures, samples with Ct>27 usually failed to deliver the full viral genome (i.e. more than >95% of the target at a minimum coverage of 5X). In addition, the reduction in coverage variation and amplicon dropouts were also dependent on the Ct value, indicating samples with Ct>27 as critical ones (Figure B).
to fragment or not to fragment
We attempted to improve our amplicon-Seq protocol for a cost-effective sequencing on NovaSeq6000 platform in PE150 mode. For this purpose, we tested an alternative library preparation protocol with the fragmentation of PCR products. The modification caused higher proportion of non-viral reads (human-derived) as compared to unfragmented amplicons, and this phenomenon was boosted for samples with low viral load (here approximated by Ct values). Bioanalyzer inspection of the amplicon traces (Figure C) suggested that non-specific amplification in highly-multiplexed PCR is, as due to stochasticity, generating products at higher molecular sizes. Thus, feeding of intact amplicons directly to the Illumina sequencer will favour the attachment of the shortest fragments resulting in a high fraction of on-target reads. On the other hand, fragmentation step will shred longer off-target products and pull them back in the “Illumina-accessible” spectrum of fragment sizes.
dna polymerase effect
Another point we found to be critical when using highly-multiplexed PCR for SARS-Cov-2 identification is the selection of DNA polymerase and its reaction buffer. Comparing NEB Q5 Hot Start DNA Polymerase mix with KAPA HiFi HotStart ReadyMix we experienced how Q5 was capable of providing a more homogeneous coverage (Figure D). This latter factor is therefore enabling reachability of nearly complete viral sequencing with lower (i.e. cheaper) average sequencing yields.
Hope you've found this post useful. Stay tuned for more updates!